Integration of epigenetic and expression platforms to understand genetic regulation in human disease
-
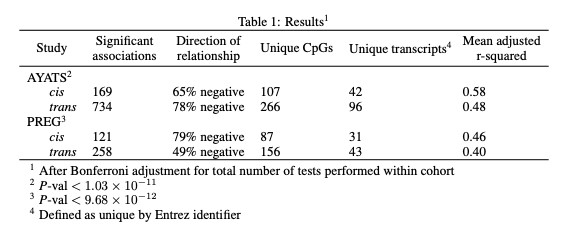
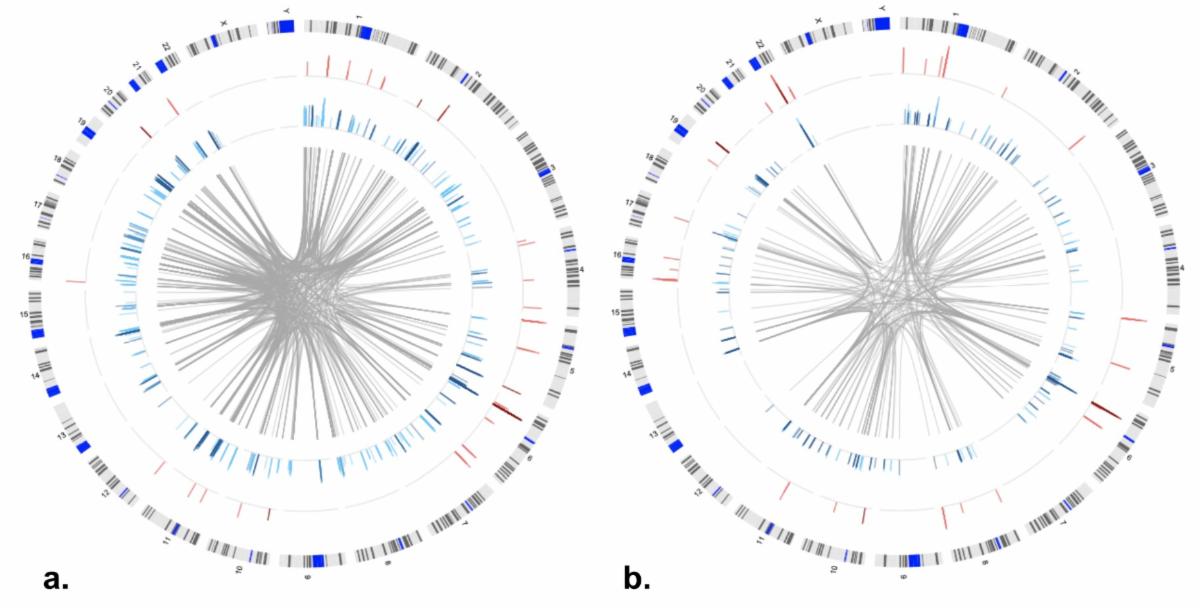
Both cis and trans relationships of large effect were identified between genome-wide measures of DNA methylation and gene expression.
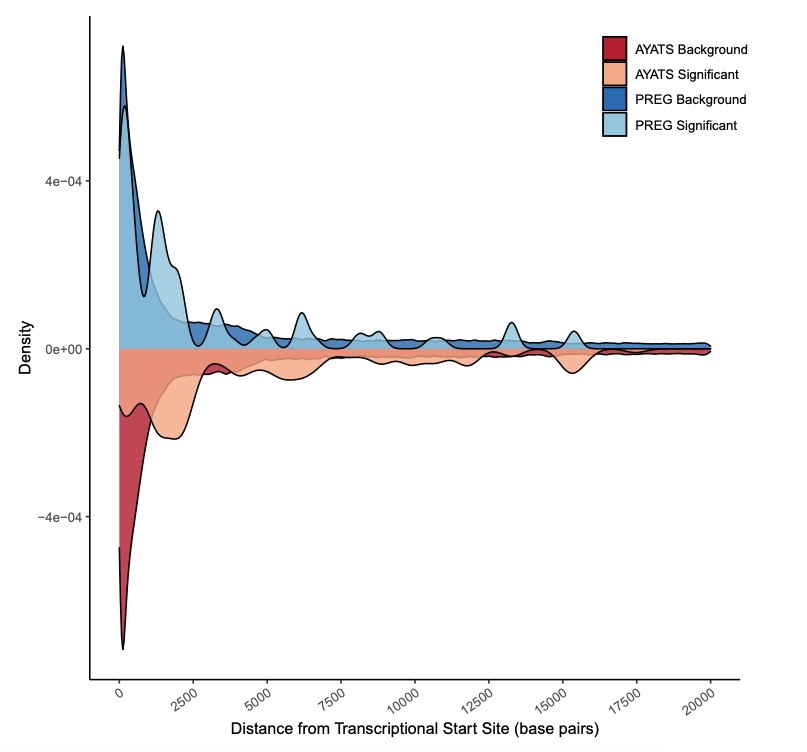
- Expression-associated CpG sites were enriched for regions of regulatory significance, including enhancer regions and transcription factor binding sites.
-
Epigenome-wide association studies should avoid assuming a canonical pathway of cis transcriptional dysregulation when interpreting disease-methylation relationships.
As an epigenetic mark, DNA methylation (DNAm) is both dynamic and persistent; modifiable by environmental exposures yet heritable during cell division. Importantly, altered DNAm has been linked to downstream functional changes, particularly in the regulation of gene expression (GE), potentially providing an explanation for how previous exposures and genetic predispositions have lasting effects on complex disease risk. Epigenome-wide association studies (EWAS) aim to test this theory that marks of DNAm have downstream consequences resulting in the development of disease. Although these methods have been successful in identifying trait-specific DNAm patterns, specific molecular mechanisms involved in the etiology of complex diseases remain unidentified. This knowledge gap does not originate from a lack of DNAm-trait associations, but rather stems from study design issues that impact the interpretability of EWAS results. Despite known limitations in predicting the function of a particular CpG site, most EWAS assume that altered DNAm results in a corresponding change in transcription at the most proximal gene. This is likely an over-simplified mechanistic interpretation that requires prior knowledge regarding the specific relationships between DNAm and GE across the genome, which has yet to be resolved.
This study aimed to improve the ability to predict downstream consequences of altered DNAm and inform best practices for interpreting EWAS results by outlining the relationships between DNAm and both proximal and distal GE (cis and trans relationships, respectively) in peripheral blood. Importantly, an in-depth functional characterization of expression-associated CpG sites was performed. To identify consistent relationships that replicate across disparate samples, this study integrated concomitant DNAm and GE measurements from two cohorts; the Adolescent and Young Adult Twin Study (AYATS; n= 137), and the Pregnancy, Race, Environment, and Genes Study (PREG; n= 131). Both cohorts measured genome-wide DNAm using the Illumina 450k HumanMethylation BeadChip (n= 485,512) and GE on the Affymetrix GeneChip Human Genome U133A 2.0 microarray (n= 14,564).
DNAm showed significant relationships with both cis (defined as within a gene region or 2.5kb upstream of the transcriptional start site) and trans (any CpG-transcript pair not otherwise categorized as proximal) GE after applying a Bonferroni correction for total number of tests performed within each cohort (Table 1). Although DNAm is usually considered a repressive mark, both positive and negative relationships between DNAm-GE pairs were identified (Figure 1). Approximately 23% of PREG connections were replicated in AYATS, with GE-associated CpG sites enriched for similar characteristics across both cohorts. CpG sites associated with GE in cis were enriched in areas of transcription factor binding and areas of intermediate-to-low CpG density. Promoter regions, which are typically characterized by high CpG density, were depleted across cohorts and within both cis and trans categories (Figure 2). DNAm exhibiting trans associations with GE were also enriched in areas of known regulatory significance, including within enhancer regions.
These results highlight issues with restricting DNAm-transcript annotations to small genomic intervals and question the validity of assuming a canonical cis DNAm-GE pathway when investigating epigenetic mechanisms. Most EWAS operate under the assumption that DNAm influences local gene transcription, but the absence of measured GE makes relying on this interpretation difficult. Based on these results, epigenetic research should incorporate an integrated multi-omic approach when studying complex traits. A modified EWAS integrating both DNAm and GE measurements still holds promise in uncovering biological processes important to the development or progression of disease, but further research outlining DNAm-GE relationships is necessary to aid in the interpretation of EWAS results that do not incorporate GE measurements.